
RNA sequencing (RNA-Seq)



ZYMO RESEARCH RELEASES OPEN-SOURCE BIOINFORMATICS PIPELIN
Total-Rna-Seq-Library-Prep


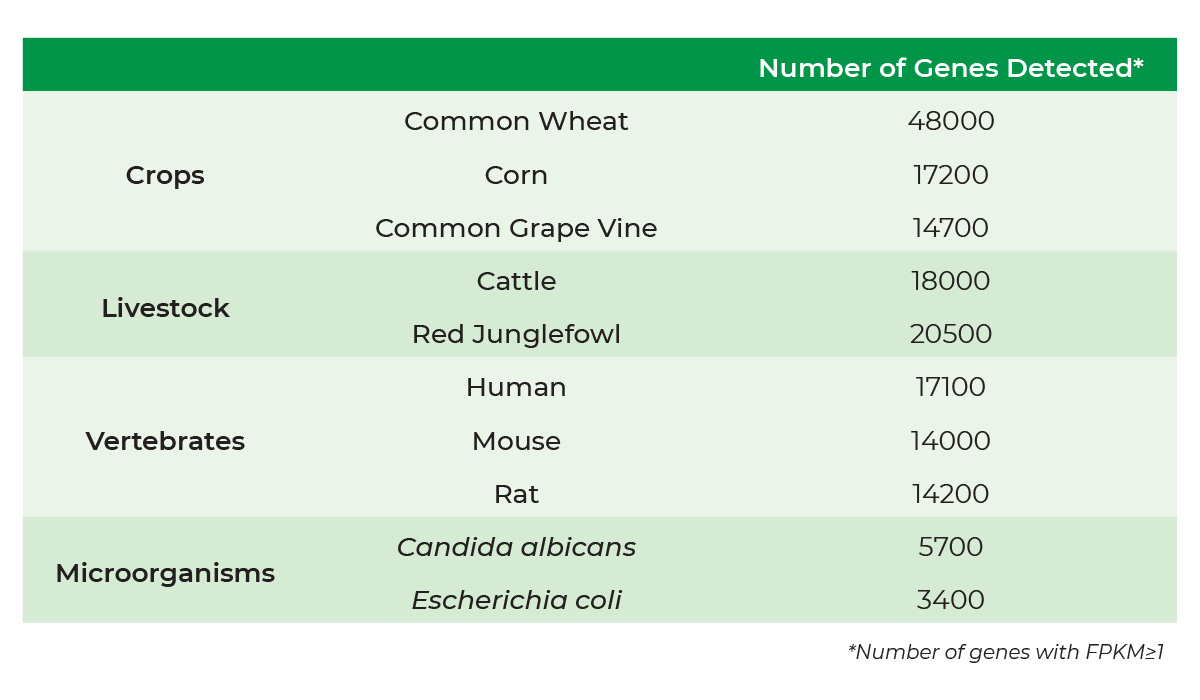
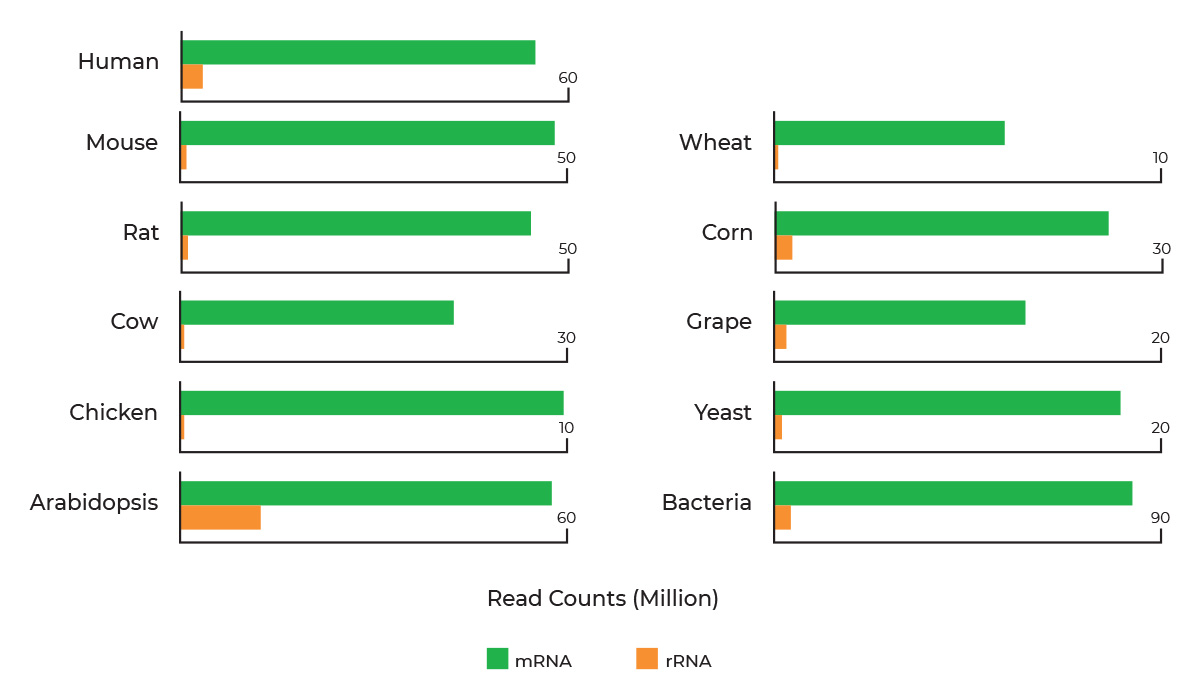
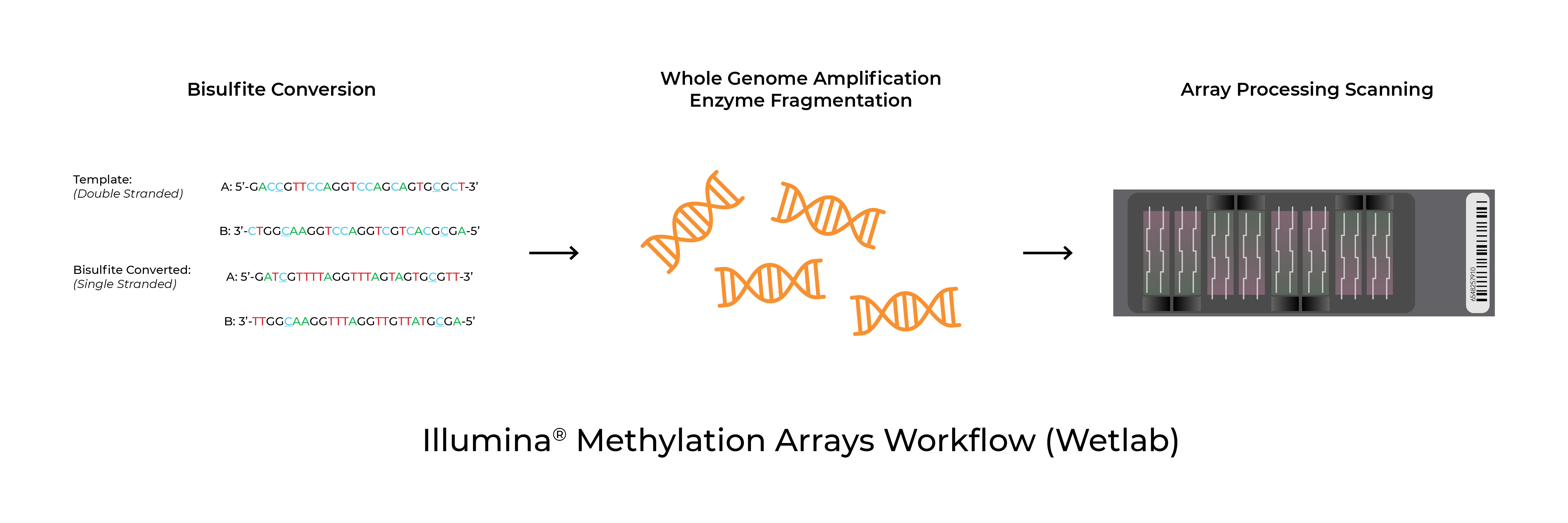
Bisulfite Conversion for Illumina Methylation Arrays
Methylation arrays are common platforms for analyzing 5-methylcytosine. The Infinium™ MethylationEPIC BeadChip and Infinium™ HumanMethylation450 BeadChip (commonly referred to as the EPIC and 450K arrays respectively) as well as the recently launched Infinium™ Mouse Methylation BeadChip from Illumina® all utilize Zymo Research’s bisulfite conversion technologies to distinguish 5mC from unmodified cytosines. In addition to the test probe sets, the arrays include bisulfite conversion quality control probes. In some instances, the analysis software will flag samples for low bisulfite conversion efficiency. Possible causes for these warnings are low bisulfite conversion efficiency, low DNA input/quality, and chip failure.
Validated Protocols
DNA Input & Quality

Low Bisulfite Conversion Efficiency

Epigenetic clock, Zymo leading the way
Is an Epigenetic Clock the Key to Asylum?
A transcript of BBC World Service Radio’s Newsday interview with Keith Booher, PhD, of Zymo Research Corporation about their DNAge® test being used to determine the age of refugees seeking asylum in Europe as minors.
BBC: Could a finger prick test for blood make it easier for child refugees to have their age verified? That’s a question that California scientists have been thinking about. The field is known as Epigenetics and the test involves examining chemically modified DNA to create a sense of how old someone actually is. Keith Booher is a scientist in California and he has been part of the development of this device.
Booher: What we’re simply trying to do is to make an age determination or quantify aging. For example, in forensics cases when the provenance or age of a sample is unknown, we can make that assessment. There’s also some indications that it may be useful for biologically aging someone – so they may have been born 45 years ago, and are 45 years old but based on certain life decision they make – the diet they choose to eat, if they get enough exercise, do they live a stress-free lifestyle? Their biological age may be older or younger than their chronological age, just depending on the lifestyle they lead. And we should be able to quantify that as well.
BBC: So if I asked you now, how old you are, Keith, what would you tell me?
Booher: I’m 38 years old.
BBC: But that’s the chronological age, is that right?
Booher: Correct!
BBC: Which one is more important, biological or chronological?
Booher: You know, if you’re a young teenager trying to get your driver’s license, obviously your chronological age is very important. But if you are in advanced years of your life or you may have made some choices that are… poor when you were younger or over the course of your life, then biological age becomes much more critical.

A Piece of the Puzzle
Epigenetic regulation – in the form of DNA methylation, histone modification, and chromatin remodelling – helps govern the proper expression of genes in the genome. One’s environment continuously shapes their epigenetics. Factors that influence epigenetics can include one’s diet, exercise, stress level, drug abuse, and exposure to toxic pollutants. Failure in epigenetic regulation can result in aberrant gene expression (mutations, deletions, copy number aneuploidies) that contributes to halted neural development 4. Thanks to progresses in Next-Gen Sequencing, scientists have been able to map over 600 human epigenes associated with neurodevelopmental disorders, including autism.

Where There’s a Whale There’s a Way
For example, the age of deceased whales can be estimated using various invasive biopsy techniques, but doing so precludes the monitoring of living subjects over time. Alternatively, researchers attempted to measure the age of live whales using telomeric genetic markers, but those methods suffer from a lack of specificity and too much technical noise. By contrast, a number of recent research articles demonstrated that DNA methylation-based age estimators, commonly referred to as epigenetic clocks, are a highly accurate and technically robust means to measure aging in mammalian species as diverse as mice, canines, humans, and other primates1-3.
A group of researchers in Australia now show that DNA methylation analysis is an effective way to help predict the age of living whale populations, thus providing a new tool to study the demographics of these majestic ocean leviathans.
In their study, Polanowski et al.4 noted the development of DNA methylation-based age clocks and set out to develop a similar test for use in humpback whale (Megaptera novaeangliae) research. They focused on the evolutionarily conserved 5’ regulatory regions of genes whose changing DNA methylation patterns correlated with age. The authors first generated a calibration data set using a cohort of 45 humpback whales originating primarily from the Gulf of Maine and with known ages ranging from a few months up to 30 years. Importantly, the tissue sample source used in the analysis came from minimally invasive skin punch dart biopsies.
