
Bisulfite PCR (BSP) is a common methodology used for methylation analysis at single-base resolution. Samples are first bisulfite converted and amplified using bisulfite-specific primers. The PCR products generated are sequenced (i.e. Next-Gen Sequencing, Sanger Sequencing, pyrosequencing) and analyzed to determine the methylation status at every cytosine. However, it is critical that primer pairs designed for BSP amplify both methylated and non-methylated sequences with the same efficiency (Figure 1). Otherwise, any amplification bias can result in skewed methylation levels.
Using methylated and non-methylated DNA controls, BSP primers can be easily optimized (validated) based on following criteria:
- +Specific product amplification
- +Equally robust amplification of both methylated and non-methylated DNA standards
- +When sequenced, the methylated and non-methylated DNA standards should result in nearly 100% and 0% methylation, respectively. If the results deviate from the expected result, this may indicate PCR bias.
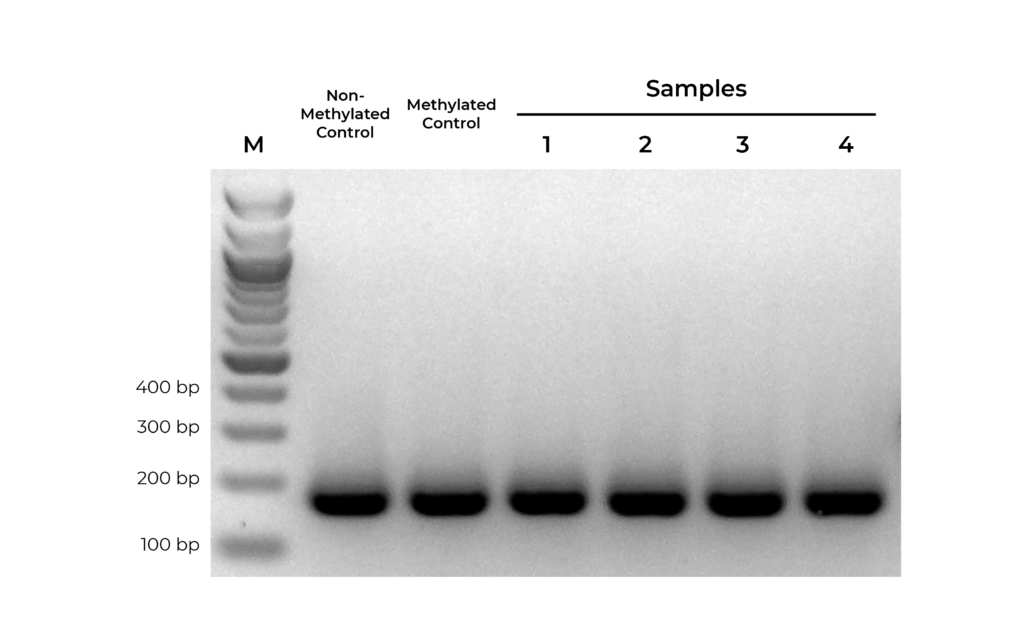
Figure 1. Bisulfite PCR primers should show equally efficient amplification of methylated, non-methylated, or partially methylated templates. Equal amounts of bisulfite converted non-methylated and methylated DNA controls were amplified in parallel with experimental samples using primers designed to target MGMT. Observed bands for methylated and non-methylated templates are of equal intensity, indicating that the primer pair is unbiased.
Unlike bisulfite PCR, methylation-specific PCR (MSP) requires methylated and non-methylated templates to be differentially amplified by two different primer sets. One is a methylation-specific primer set, and if amplification occurs, it indicates the amplified region is completely methylated. The second primer set targets non-methylated templates, so any amplification with this primer set indicates the amplified region is completely non-methylated. If amplicons are generated by both primer sets, the region is partially methylated.
Such two primer sets required for MSP can also be optimized and validated utilizing Methylated & Non-Methylated DNA Controls using following guidelines:
- #Specific product amplification
- #The methylated primer will only amplify the methylated control DNA and will not amplify the non-methylated DNA.
- #The non-methylated primer will only amplify the non-methylated control DNA and will not amplify the methylated DNA.
During primer validation, having characterized methylated and non-methylated DNA standards can streamline the process. For example, if a slight PCR signal is observed using the methylated standard with the non-methylated primers, or vice versa, it would indicate that the primer set is non-specific. For established workflows, especially in clinical assays, methylated and non-methylated DNA standards can serve as positive and negative controls to validate the MSP workflow. Standards will help confirm that the entire process – bisulfite conversion, amplification, and analysis – are functioning appropriately, and the results for the test samples are reliable (Figure 2).
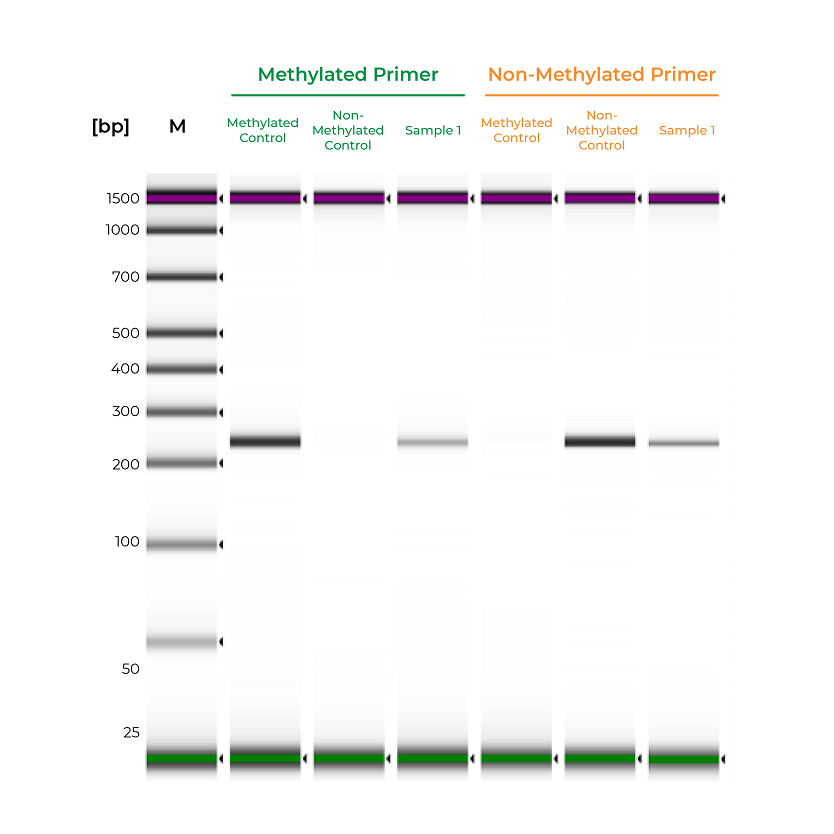
Figure 2. Methylated and non-methylated controls are important for validating the sensitivity of MSP assays. The methylated and non-methylated controls are only amplified by the methylated and non-methylated primers, respectively. Sample 1 is amplified by both primer sets, indicating mixed methylation. Samples were analyzed on the Agilent D100 ScreenTape.
In addition to validating primers, DNA standards can be used in place of precious samples to optimize the annealing temperature for new primer sets (Figure 3). An annealing temperature gradient should be performed for each newly designed primer set to ensure optimal amplification of the intended target.
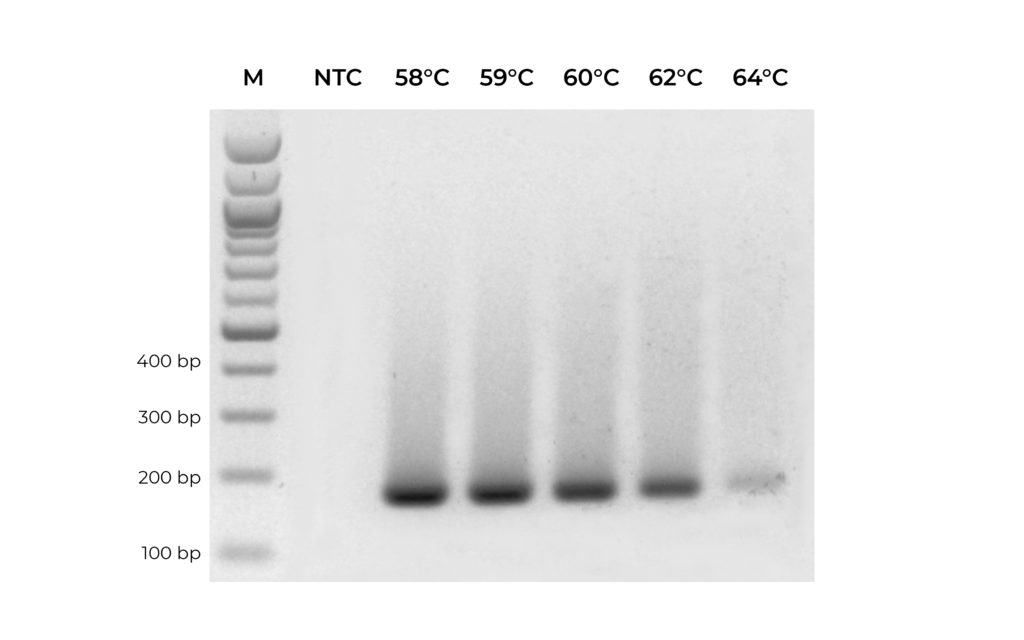
Figure 3. A primer set targeting MGMT was optimized by testing replicate PCR reactions with annealing temperature ranging from 58°C to 64°C.
Methylation sensitive restriction enzyme (MSRE) assays rely on restriction enzymes being able to distinguish methylated versus non-methylated cytosines. If the restriction site is not methylated, the enzyme will digest the DNA while methylated sites will be uncut. Then, methylation at the site can be evaluated using quantitative PCR by comparing the amplification of the digested versus undigested sample. The smaller the ∆C(t) between the two, the higher the methylation levels are, and vice versa (Figure 4). DNA visualization methods, such as an agarose gel, can also be used to determine if the digestion occurred at the region of interest.
Methylated and non-methylated DNA standards should be digested in parallel to experimental samples to demonstrate the robustness of the restriction enzyme in distinguishing methylated versus non-methylated sites. This will also help determine the sensitivity of the assay to the region of interest.
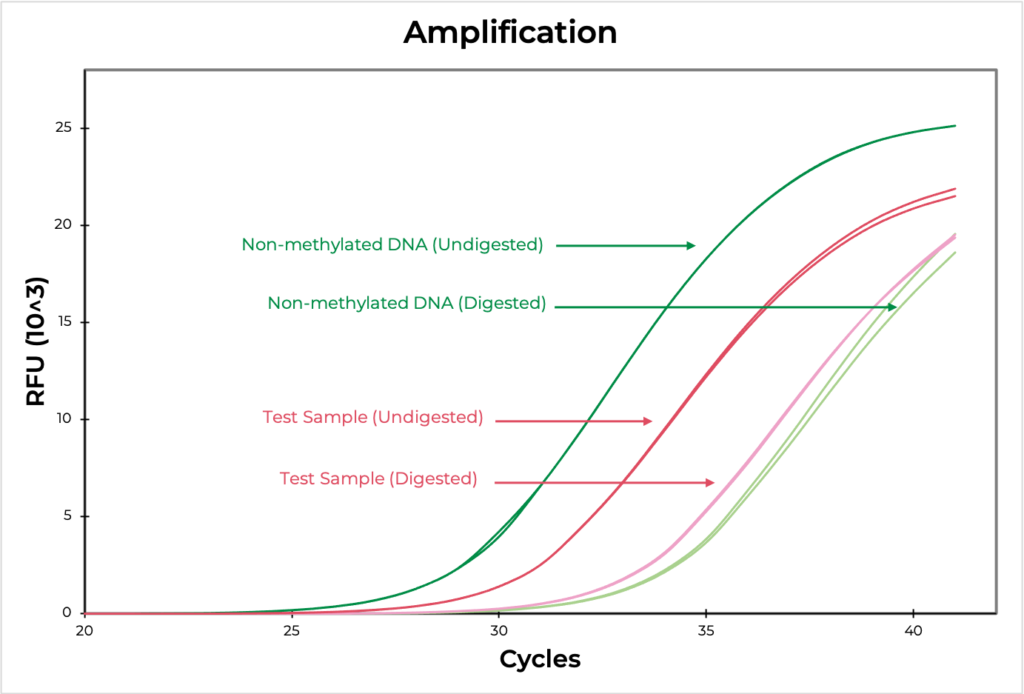
Figure 4. The amplification curve demonstrates the differences in Ct values between digested and undigested samples. The MSRE assay detected 100.9% and 2.7% methylation in the methylated and non-methylated DNA standards, respectively, confirming that the assay worked efficiently. The test sample methylation level was determined to be 17.3.