There is common perception that rRT-PCR tests detecting multiple SARS-CoV-2 gene targets perform better than those using a single SARS-CoV-2 target. However, it is important to understand that the number of targets detected by an assay is not an indication of test sensitivity or performance. In fact, a study published in Nature Biotechnology compared over 150 rRT-PCR tests authorized by the American FDA and found that over 25% of the tests were designed using a single viral target 7. In the same study, the top six tests with the lowest limit of detection (highest sensitivity) used exactly one SARS-CoV-2 target 7. This indicates that detecting a single SARS-CoV-2 target does not reduce test performance, as many of the most sensitive tests on the market actually only detect one viral target.
One reason behind the common misconception that it is necessary to have multiple SARS-CoV-2 targets for rRT-PCR is the concern that the virus could mutate in a binding site for the primer and/or probe. This event would ultimately invalidate a single viral target test, but not a multi-target test. Although possible, this is very unlikely to happen, particularly if the selected single viral target is classified as “stable.”
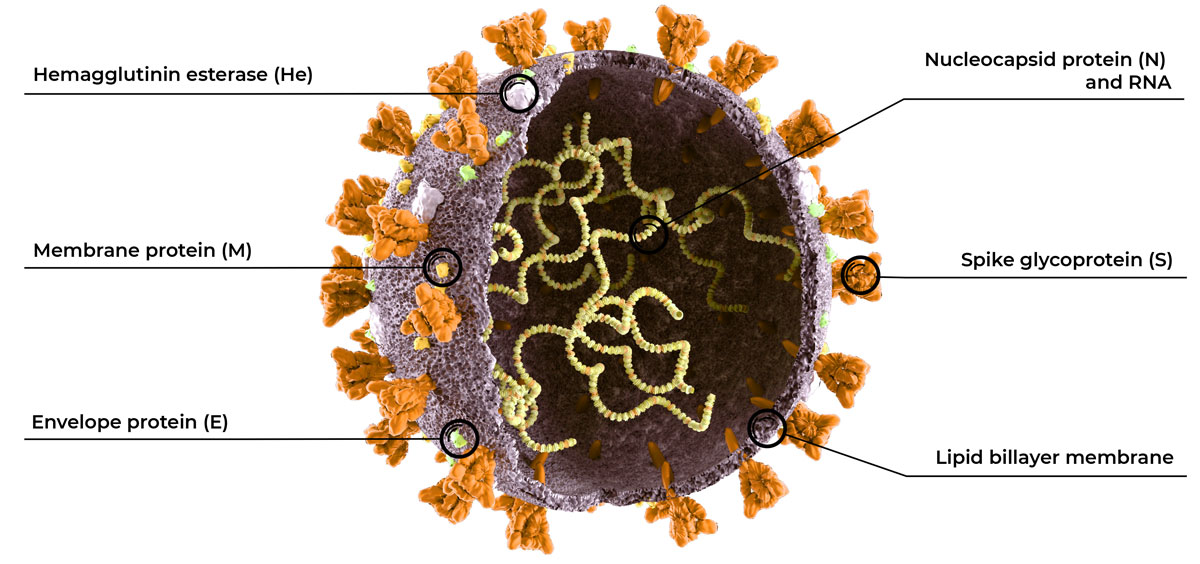
This is particularly true for coronaviruses. This virus family possesses genomes of 26 to 32 kb in length, which is larger than many other RNA viruses (in comparison, HIV and Influenza A genomes are 9.2 and 13.6 kb in length, respectively). Viruses with larger genomes experience higher fitness costs associated with replication errors and mutations8. To compensate for this, coronaviruses (unlike most virus families) use the nonstructural protein 14 (nsp14) to remove misincorporated ribonucleotides before a new RNA strand is extended 8.
The result is that coronaviruses make far fewer replication errors than other RNA viruses, and therefore have fewer mutations and less genetic variability over time 8. For example, it has been estimated that SARS-CoV-2 mutates at a rate that is approximately one-half that of influenza and one-quarter that of HIV 9. Because SARS-CoV-2 is mutating much slower than other RNA viruses, it is not as critical to develop diagnostic testing that prepares for the chance of mutation.
Furthermore, in order to invalidate a single-target rRT-PCR test, a mutant viral strain would need to become established among the population. This is only possible if the mutation confers an evolutionary advantage to the virus. An example is the new SARS-CoV-2 variant that was first detected in the UK in December 2020 and is now spreading worldwide 6. This SARS-CoV-2 variant has multiple mutations in the S gene (which encodes the spike protein) that increase the transmissibility of the strain, which is a clear evolutionary advantage. Most mutations, however, are deleterious or neutral, meaning their frequency in the population will remain negligible and they will not compromise the performance of single-target rRT-PCR tests, especially those designed on a genetically stable region like certain parts of the N gene.
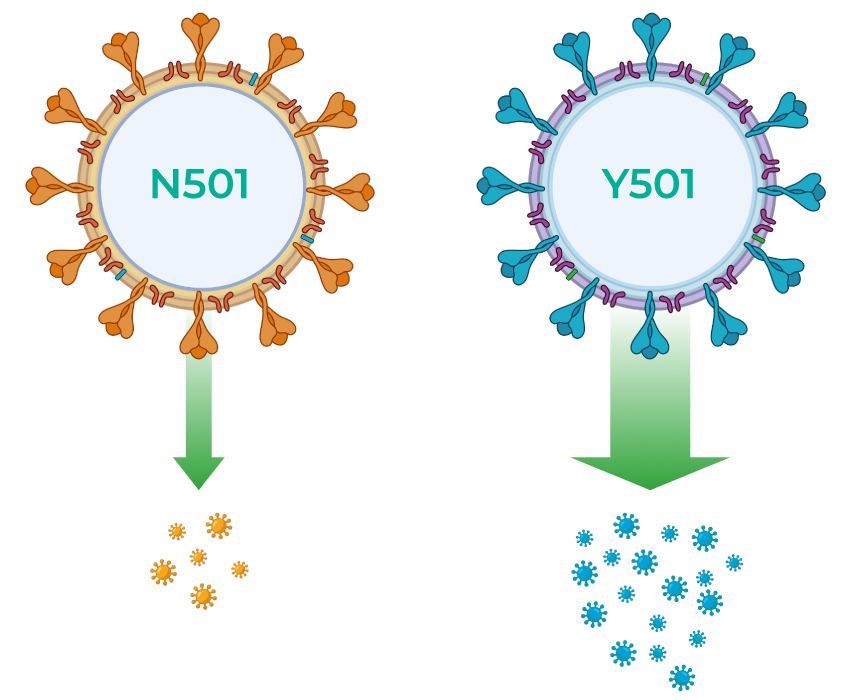
FIG: The variant strain of SARS-CoV-2 that first became prevalent in the UK contains multiple mutations in the spike (S) protein, one of which is an amino acid change from asparagine (N) to tyrosine (Y) at position 501 in the receptor-binding domain of the protein10. The spike protein is under strong selective pressure because it directly affects the transmissibility of the virus.