The paper strip test was developed at the Council of Scientific and Industrial Research’s constituent lab, the Institute of Genomics and Integrative Biology (CSIR-IGIB) based in New Delhi, India. The testing kit addresses the urgent need for accurate COVID-19 mass testing and has many advantages compared to the gold standard, qPCR. Like SHERLOCK (specific high-sensitivity enzymatic reporter unlocking) which is a COVID 19 test using Cas12 for detection of SARS-CoV-2, developed by scientists from the Broad Institute in the US, the name Feluda refers to an Indian fictive detective (film director: Satyajit Ray).
Advantages of the Feluda test compared to qPCR:
# Affordability
# Can be used in settings with limited resources
# Less time to result (around 45 minutes)
# No qPCR equipment (no need for expensive devices)
# High ease of use
# Lateral Flow based readout for easy interpretation of results
With 96% sensitivity and 98% specificity, Feluda’s analytical performance is comparable to the qPCR results.
How the Feluda test works
The COVID-19 test developed in New Delhi, India by a research team led by Debojyoti Chakraborty and Souvik Maiti is based on a Cas9 enzymatic readout for detecting nucleotide sequences and identifying nucleobase identity. Just a few weeks ago, Emmanuelle Charpentier and Jennifer Doudna have been awarded the 2020 Nobel Prize in Chemistry for their development of CRISPR/Cas9 gene editing technology. The Cas9 readout used for coronavirus detection has requirement of trans-cleavage activity of reporter molecules like Cas12 or Cas13 methods.
The scientists from CSIR-IGIB used Cas9 from Francisella novicida (FNCas9), which shows very high mismatch sensitivity and can distinguish between nucleotide sequences differing by only one mismatch. The principle of the paper strip test was originally designed for the identification of sickle cell anemia, a disease caused by a point mutation, and adapted to COVID-19 testing due to the Coronavirus outbreak resulting in an urgent need for mass testing.
The FnCas9 used in this new method does not produce collateral activity on substrates due to the use of a catalytically inactive FnCas9-gRNA-complex. Therefore, it is an affinity based method and no trans-cleavage signal output is generated.
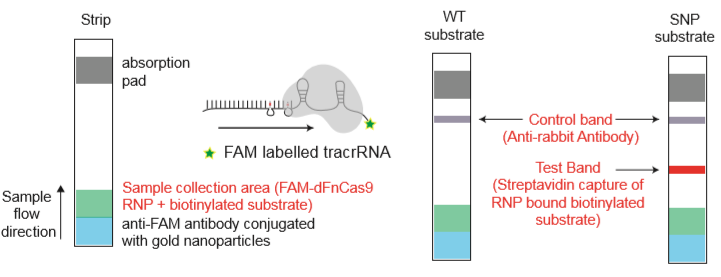
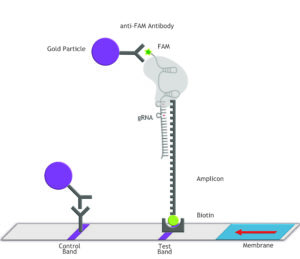
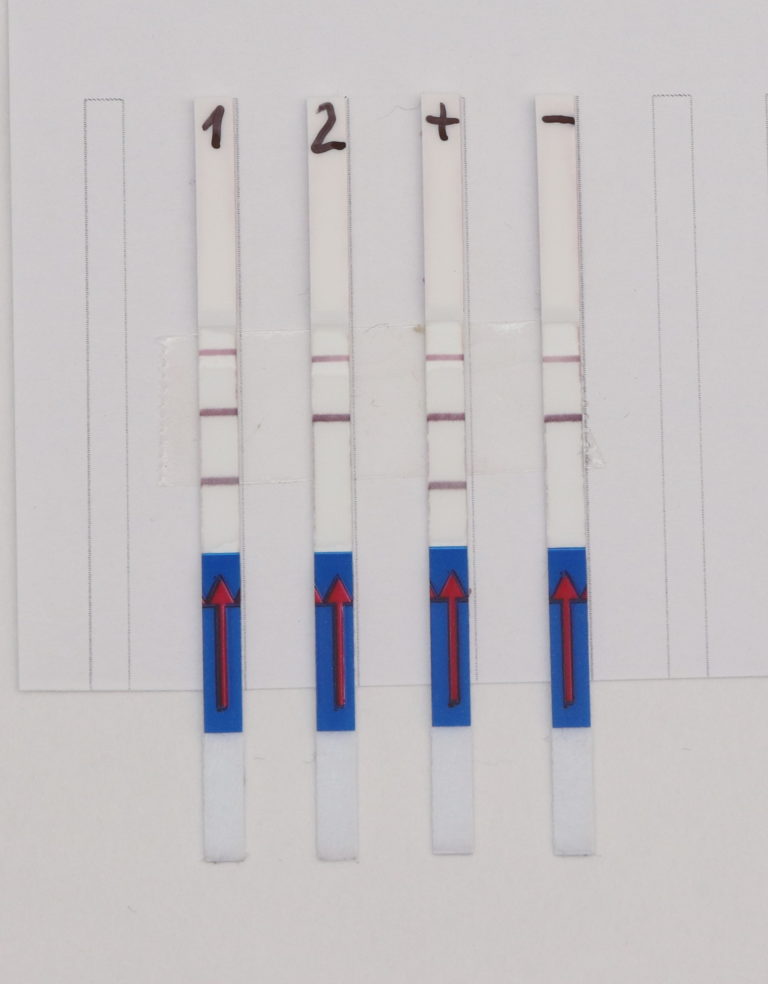
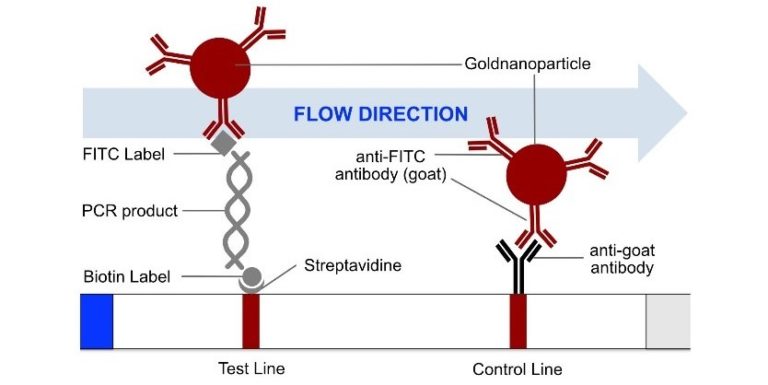
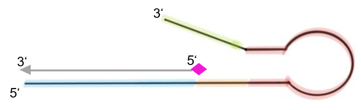
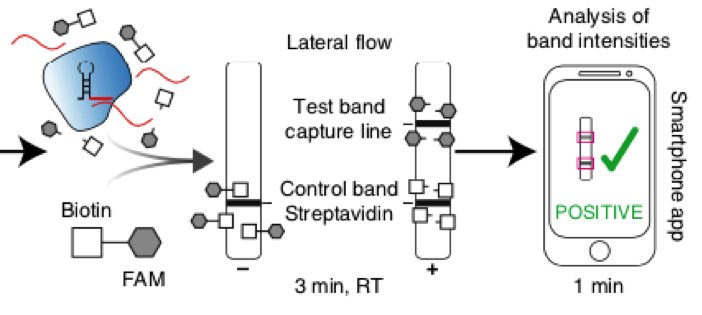
Fig. 1: Feluda test principle on HybriDetect. FnCas9 with FAM labeled gRNA binds to biotinylated sample if result is positive.
The Feluda test and Milenia HybriDetect
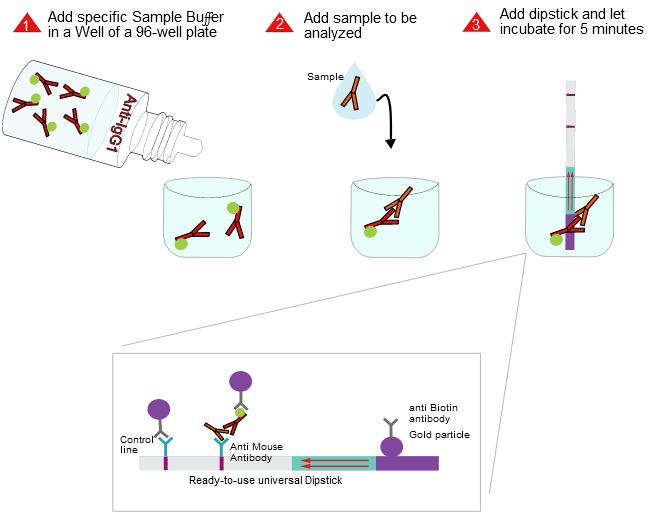
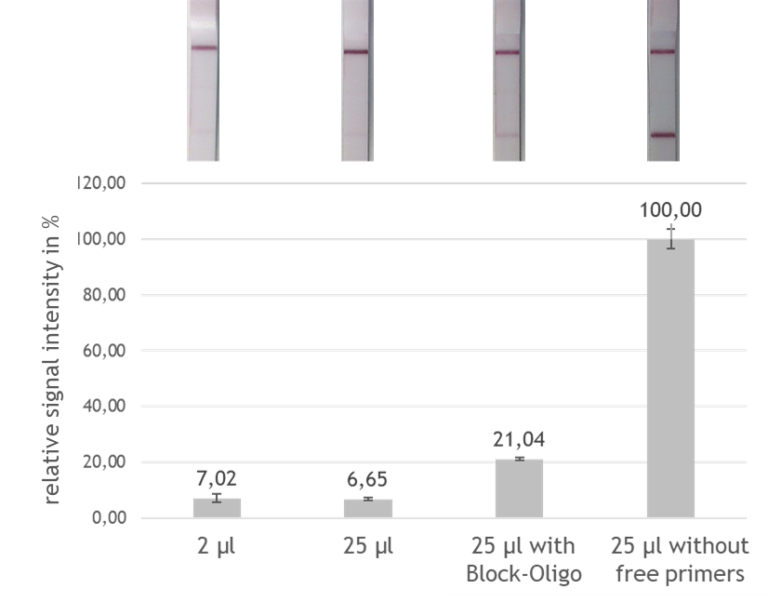
The indian researchers designed a gRNA, labeled with FAM, which is important for the visualization of the test line on Milenia HybriDetect (Figure 2).
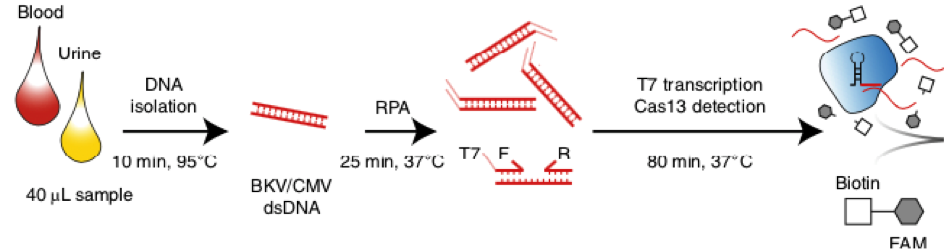
After RNA extraction, the first step of the paper strip test is an optimized single step Reverse Transctription-PCR (RT-PCR), or alternatively a RPA protocol, where the sample gets amplificated and biotinylated.
In the next step, the FnCas9 gRNA (labeled with FAM) is incubated with the biotinylated substate (virus sequence if sample is positive). Due to the streptavidin, immobilized on the HybriDetect dipstick, the RNP Complex (CRISPR ribonucleoprotein) binds to the test line (Figure 2).
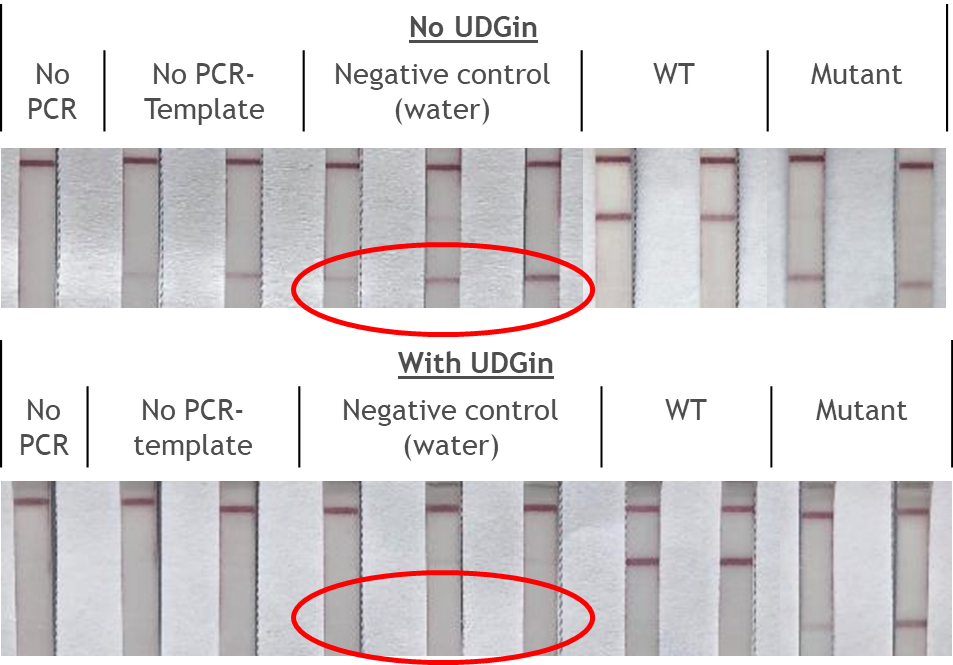
The RNP-complex bound to the labeled substrate is visualized with anti-Fam antibodies conjugated to gold nanoparticles and a positive test line is formed if substrate is bound to the RNP-complex. Summarized this means, two lines for a positive and a single line for a negative test result. The visual interpretation of the HybriDetect dipstick is as easy as the interpretation of a pregnancy test and therefore no trained personnel is required.
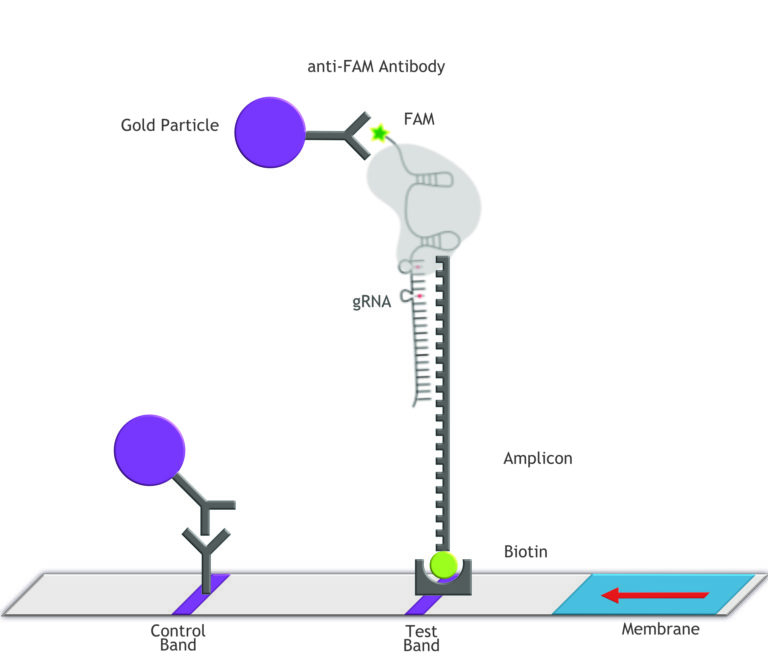
Fig.2: Milenia HybriDetect and FnCas9. The test is positive if the biotinylated DNA is present and binds to streptavidin on the T-Line. FnCas9 binds to the biotinylated DNA and is captured by FAM antibodies coated with gold nanoparticles for visualization.
When to expect Feluda Test to be available?
The Union Health Minister of India Harsh Vardhan said on October 11th, that the rollout of the new Covid-19 test is expected in the next few weeks. The test developed by scientists from the Council of Scientific and Industrial Research’s Institute of Genomics and Integrative Biology in New Delhi is going to provide results in 45 minutes and is priced around Rs 500 ( ~ 5,70 Euro).
Additional Information on the Feluda test
# Compared to other CRISPR methods for COVID Detection, including SHERLOCK and DETECTR, Feluda does not need a reporter and is therefore less complex.
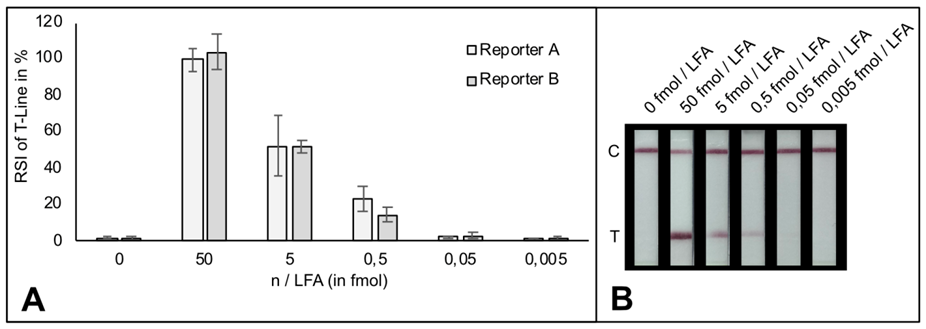
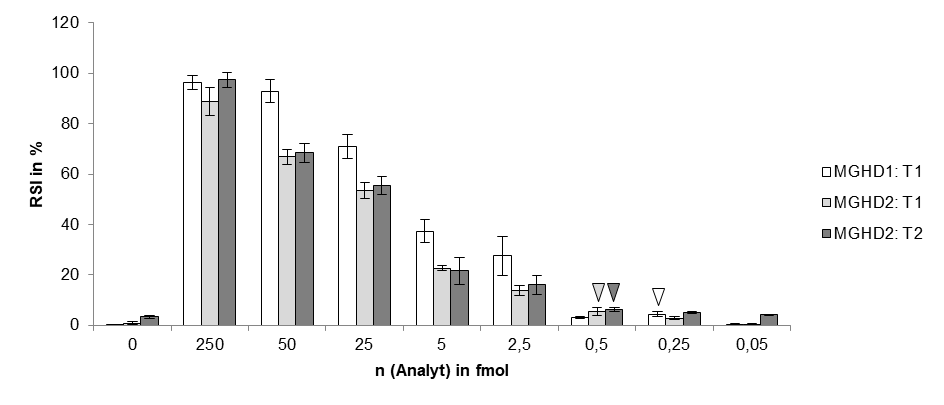
# The LOD of indias new test method is 10 copies of purified viral sequence as the authors mention in their latest publication (https://www.medrxiv.org/content/10.1101/2020.09.13.20193581v1.full.pdf)
# To assist the detection of COVID-19 the scientists developed a smartphone app named TOPSE (True Outcome Predicted via Strip Evaluation) which gives a predictive score based on background correction. Due to the stoichiometric binding affinity of FnCas9 RNP to the target, a semi-quantitative readout of Feluda is possible with TOPSE.
Feluda for at home testing
The Scientists from CSIR-IGIB are currently working on a Feluda version for at home coronavirus testing. Therefore they are trying to do a machine free, RPA based amplification of the virus genome. The aim is an end-to end instrumentation free testing protocol.